近日,我校生命科学学院计算与整合生物学团队在国际基因组学领域权威学术期刊《Genome Biology》(IF=12.3,生物学一区Top)上在线发表了题为“Systematic evaluation with practical guidelines for single-cell and spatially resolved transcriptomics data simulation under multiple scenarios”的研究论文,对团队在单细胞RNA测序和空间转录组学方法学领域取得的重要进展进行了介绍。该研究全面评估了单细胞/空间转录组数据模拟算法在多个流行应用场景下的表现,并为该领域的科研人员提供了详尽的方法选择使用指南。

bat365官网登录入口为论文第一完成单位和唯一通讯单位,生命科学学院2022级硕士研究生朵泓睿为第一作者,李勃副教授和郝友进教授为通讯作者,重庆邮电大学、陆军军医大学、浙江大学、重庆大学和重庆医科大学等单位的研究人员共同参与了此项工作。
近年来,随着单细胞RNA测序(scRNA-seq)和空间转录组学(SRT)的迅速兴起,两者通过精确揭示单细胞水平基因表达的异质性及其在组织中的空间分布特征,显著加速了现代生命科学的研究进程,推动了研究范式的转变和创新。在scRNA-seq和SRT算法设计和数据分析工具基准测试中,模拟数据至关重要。尽管目前已有大量用于模拟scRNA-seq和SRT数据的方法,但面对具体的生物信息学分析任务时,如何选择最适合的方法仍然是一个棘手的问题。
在该项研究中,团队成员利用152组真实参考数据集对49种单细胞/空间转录组数据模拟方法在准确性、功能性、可拓展性和适用性方面开展了大规模的系统性评估。研究结果表明,建立在最优选择模型(e.g., SRTsim 和 scDesign2)和 GAMLSS(e.g., scDesign3)基础上的方法具有最佳的准确性表现,但需要以消耗更多时间和内存来建立基因表达数据模型为代价。此外,ZINB- WaVE、SPARSim、Splat、SCRIP-paths、muscat 和 SCRIP-GP-trendedBCV 也表现出较强的数据模拟能力。专用于scRNA-seq数据模拟的方法Splat、SPARSim、SCRIP、SplatPop、dropim 和 ZINB-WaVE 在模拟 SRT 数据方面具有强大的兼容性。在功能性方面,Lun在模拟细胞类群和差异表达基因的应用场景下的效果最佳,而 SPARSim和scDesign3-tree 则分别在细胞批次和轨迹模拟方面表现优于其他方法。由于没有一种方法在所有评价标准上都表现出色,因此用户应考虑在准确性和功能性、准确性和可扩展性之间做出权衡。在适用性方面,半数以上的方法在运行过程中会产生错误,而且错误比例在不同方法间具有较大差异。“基因表达值拟合失败”和 “出现缺失(无穷)值 ”是运行失败的两个主要原因。
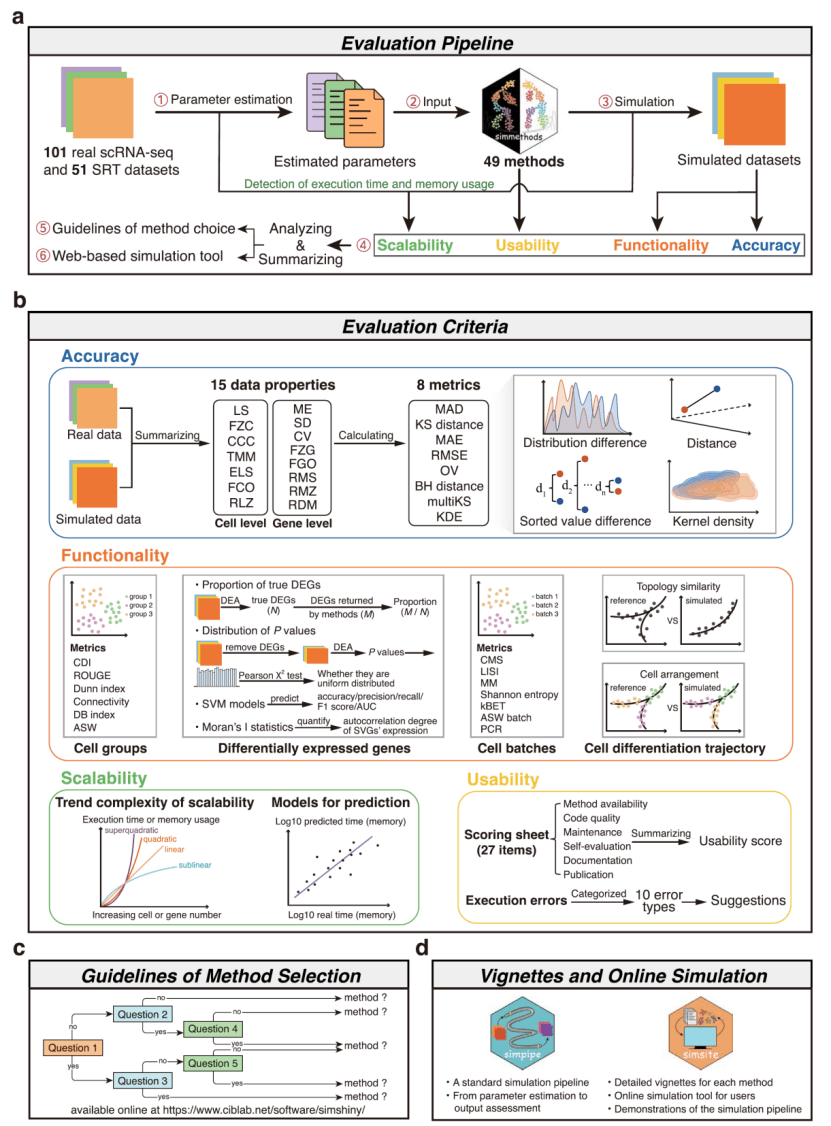
根据评估结果,团队成员还建立了方法选择的实用指南、标准数据模拟流程Simpipe和交互工具 Simsite(https://www.ciblab.net/software/Simsite/),以供用户选择合适的方法并执行模拟任务。这项研究将为单细胞组学领域的研究者提供指导和建议,帮助他们选择合适的数据模拟方法。同时,它也将激励算法开发人员提出更具扩展性和效率的方法,从而有助于研究者深刻理解和应用基因表达数据的特征。
据悉,《Genome Biology》是国际基因组生物学领域顶级学术期刊,主要发表从基因组和后基因组的角度研究生命科学的最新研究成果,创刊以来刊登过多项重大的生命科学研究进展,年刊载文章数为200-300篇。该期刊最新影响因子12.3,其5年影响因子为17.4,目前为中科院一区TOP期刊。
原文链接


 渝公网安备 50009802500172号
渝公网安备 50009802500172号

